Scientific Corner
Looking for our published papers? The Scientific Corner is the place for you.
Scientific Publications and Posters
+
Discover What’s New










Papers in Peer Reviewed Journals
+
Once-daily Oxycodone prolonged-release tablets are resistant to alcohol-induced dose dumping: results from a randomised trial in healthy volunteers. Journal of Pain, 2019
Rayad N, Wong W et al. Antiviral treatment for treatment-naïve chronic hepatitis B: systematic review and network meta-analysis of randomized controlled trials (August 2019)
Trabelsi F, Gharavi N, Kalovidouris M, Nikolaidou M. Steady-state Bioequivalence study of two quetiapine prolonged-release 400 mg tablet formulations in healthy male and female subjects under fasting. Int J Clin Pharmacol Ther. 2016,;54:732-742.
Assessment of the abuse potential of the Orexin receptor antagonist, Suvorexant, compared with Zolpidem in a randomized crossover study.Journal of Clinical Psychopharmacology, 36(4):314-323, Aug 2016
Assessment of pharmacokinetic and pharmacodynamics interactions between albumin-fused mutated butyrylcholinesterase and intravenously administered cocaine in recreational cocaine users. Journal of Clinical Psychopharmacology, 35(4):364-366, Aug 2015
Assessment of Pharmacokinetic and Pharmacodynamic interactions between albumin-infused mutated butyrylcholinesterase and intravenously administered cocaine in recreational cocaine users, Journal of Clinical Psychopharmacology (2015), 36(4):314-323
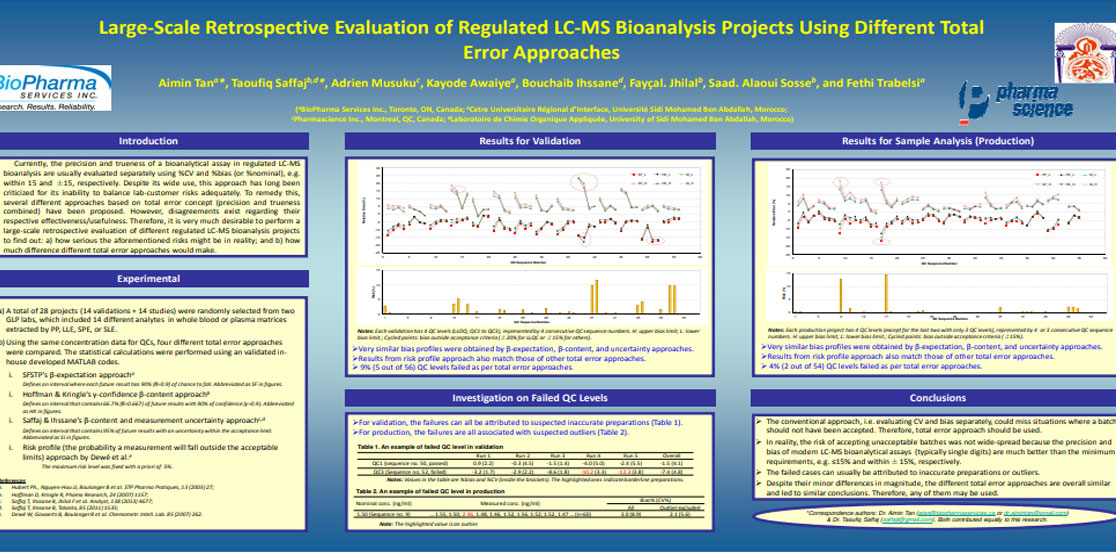
Aimin Tan, Taoufiq Saffaj, Adrien Musuku, Kayode Awaiye, Bouchaib Ihssane, Fayçal Jhilal, Saad. Alaoui Sosse, Fethi Trabelsi. Large-Scale Retrospective Evaluation of Regulated Liquid Chromatography-Mass Spectrometry Bioanalysis Projects Using Different Total Error Approaches. Journal of Chromatography B, 983-984 (2015) 68-75.
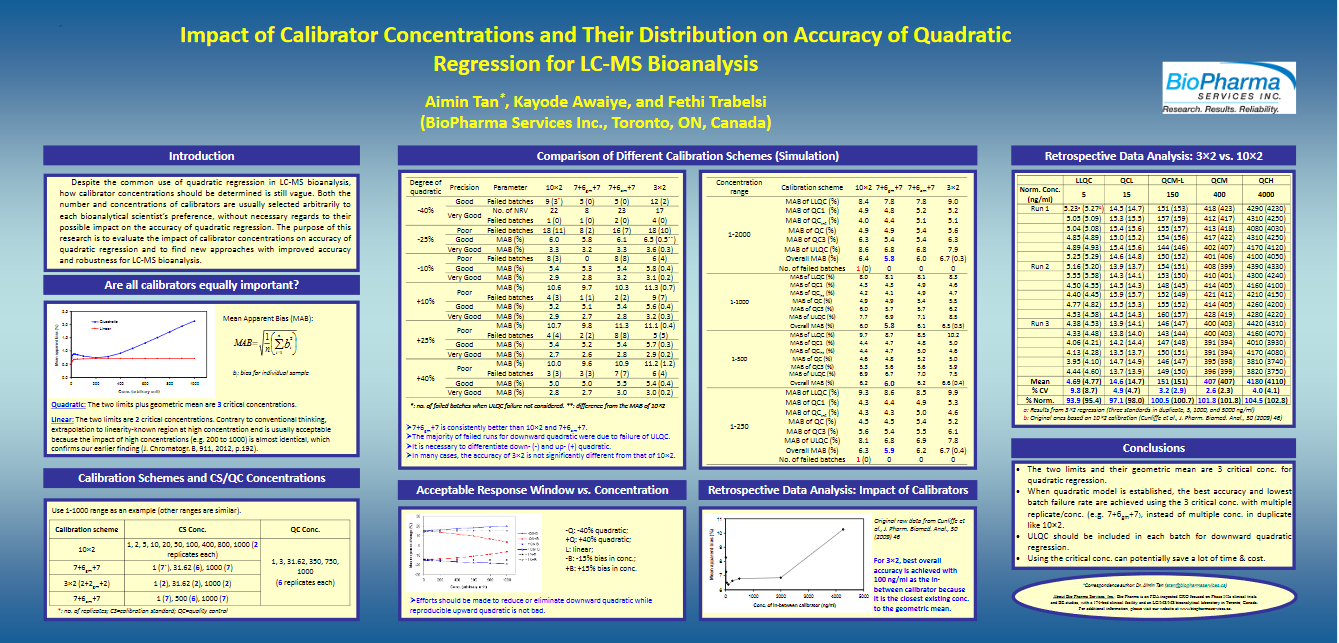
Tan, Aimin; Awaiye, Kayode and Trabelsi, Fethi. Impact of calibrator concentrations and their distribution on accuracy of quadratic regression for LC-MS Bioanalysis. Anal Chim Acta. (2014) Mar 7; 815: 33-41.doi: 10.1016/j.aca.2014.01.036. Epub 2014 Jan 19.
Musuku A., A. Tan, K. Awaiye, F Trabelsi. Comparison of two-concentration with multi-concentration linear regressions: Retrospective data analysis of multiple regulated LC-MS bioanalytical projects. J. Chromatography B. 934 (2013), 117-123.
“Differences in the Vitro and In Vivo Pharmacokinetic Profiles of Once-Daily Modified-Release Methylphenidate Formulations in Canada: Examination of Current Bioequivalence Criteria.” Clinical Therapeutics, Apr 2012
Relative abuse potential and safety of a new formulation Oxycodone HCl tablets (Orexta) in non-dependent opioid users. Journal of Opioid Management, 8(5):315, 2012
Assessing subjective and physiologic effects following intranasal administration of a new formulation of immediate-release oxycodone HCI (OxectaTM) tablets in nondependent recreational opioid users. Journal of Opioid Management, Sep 27, 2012
AiminTan, Kayode Awaiye, Besy Jose, Paresh Joshi, and Fethi Trabelsi. Comparison of Different Linear Calibration Approaches for LC-MS Bioanalysis. Journal of Chromatography B, 911 (2012), 192-202.
“Intranasal Abuse Potential of an IR Oxycodone Formulations (Poster Presentation).” American Academy of Pain Management 22nd Annual Clinical Meeting, Sep 23, 2011
“Intranasal Abuse Potential of Immediate-Release Oxycodone (Accurox, IROA) Formulated to Deter Abuse (Poster Presentation).” College on Problems of Drug Dependence, Jun 2011
Publications
+
Assay List
+
Edited by: JeanNicholas Mess, Qin C Ji, Dafang Zhong, Long Yuan, and Annik Bergeron
Publisher:
Publication Date: October, 2015
________
Aimin Tan and Kayode Awaiye, Chp 17: Use of Internal Standard in LC-MS Handbook of Bioanalysis, Best Practices, Experimental Protocols and Regulations.
Edited by: Wenkui Li, Jie Zhang and Francis LS Tse
Publisher:John Wiley
Publication Date: October, 2013
Schedule a Discovery Call
You can unsubscribe at any time. For more details, please read our Privacy Policy.